
TIVO-3: a study in renal cell carcinoma (RCC) patients after 2 or more systemic therapies

TIVO-3: a study in renal cell carcinoma (RCC) patients after 2 or more systemic therapies
TIVO-3 is the first and only prospective, Phase 3 study of a VEGFR TKI in rrRCC after 2 or more systemic therapies including IO1,2
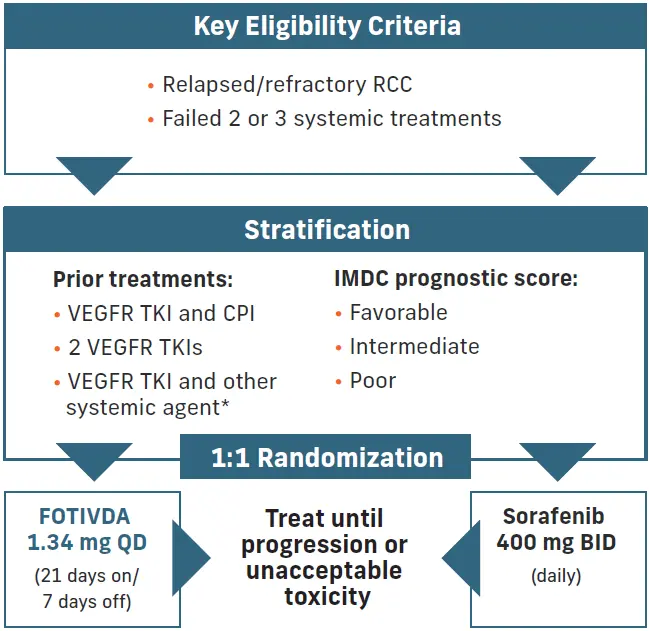

Study design: Open-label, multicenter study (N=350) in North America and the European Union
Primary endpoint: PFS assessed by a blinded independent radiology review committee
Secondary endpoint: ORR (CR+PR), DOR, OS, tolerability and safety
*Other systemic agents were mTOR inhibitors and cytokines (interferon and interleukin).2
Get an overview of the TIVO-3 study design and its uniqueness within the RCC treatment paradigm from Dr. Pedro C Barata, Director, GU Medical Oncology Research Program at University Hospitals Seidman Cancer Center.
View a video of Dr. Barata covering the importance of the first and only Ph3 data after 2 or more prior regimens that reflects how providers manage RCC patients in the real-world.
Get an overview of the TIVO-3 study design and its uniqueness within the RCC treatment paradigm from Dr. Pedro C Barata, Director, GU Medical Oncology Research Program at University Hospitals Seidman Cancer Center.
View a video of Dr. Barata covering the importance of the first and only Ph3 data after 2 or more prior regimens that reflects how providers manage RCC patients in the real-world.
See results from the pivotal trial.
Explore safety and tolerability.
Reach out to an AVEO Oncology Account Manager.
BID=twice daily; CPI=checkpoint inhibitor; CR=complete response; DOR=duration of response; IMDC=International Metastatic RCC Database Consortium; IO=immuno-oncology; mTOR=mechanistic target of rapamycin; ORR=overall response rate; OS=overall survival; PR=partial response; PFS=progression-free survival; QD=once daily; rrRCC=relapsed/refractory renal cell carcinoma; TKI=tyrosine kinase inhibitor; VEGFR=vascular endothelial growth factor receptor.
References: 1. FOTIVDA (tivozanib) (package insert], Boston, MA: AVEO Pharmaceuticals, Inc, January 2025. 2. Rini BI, Pal SK, Escudier BJ, et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. 2020;21(1):95-104.
INDICATIONS
FOTIVDA is indicated for the treatment of adult patients with relapsed or refractory advanced renal cell carcinoma (RCC) following two or more prior systemic therapies.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hypertension was reported in 45% of patients (22% ≥ Grade 3). Hypertensive crises were reported in 0.8% of patients. Do not initiate FOTIVDA in patients with uncontrolled hypertension. Monitor for hypertension and treat as needed. Reduce the FOTIVDA dose for persistent hypertension not controlled by anti-hypertensive medications. Discontinue FOTIVDA for severe hypertension that cannot be controlled with anti-hypertensive therapy or for hypertensive crisis.
INDICATIONS
FOTIVDA is indicated for the treatment of adult patients with relapsed or refractory advanced renal cell carcinoma (RCC) following two or more prior systemic therapies.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hypertension was reported in 45% of patients (22% ≥ Grade 3). Hypertensive crises were reported in 0.8% of patients. Do not initiate FOTIVDA in patients with uncontrolled hypertension. Monitor for hypertension and treat as needed. Reduce the FOTIVDA dose for persistent hypertension not controlled by anti-hypertensive medications. Discontinue FOTIVDA for severe hypertension that cannot be controlled with anti-hypertensive therapy or for hypertensive crisis.
Cardiac failures were reported in 1.6% of patients (1% ≥ Grade 3); 0.6% of events were fatal. Monitor for signs or symptoms of cardiac failure during treatment with FOTIVDA. Manage with dose interruption, dose reduction, or discontinuation.
Cardiac ischemia were reported in 3.2% of patients; 0.4% of events were fatal. Arterial thromboembolic events were reported in 2.0% of patients, including death due to ischemic stroke (0.1%). Closely monitor patients at risk for, or who have a history of these events. Discontinue FOTIVDA in patients who develop severe arterial thromboembolic events, such as myocardial infarction and stroke.
Venous Thrombotic Events (VTE) were reported in 2.4% of patients, including 0.3% fatal events. Closely monitor patients who are at increased risk for these events. Discontinue in patients who develop serious VTEs.
Hemorrhagic Events were reported in 11% of patients; 0.2% of events were fatal. Use FOTIVDA with caution in patients who are at risk for or who have a history of bleeding.
Proteinuria was reported in 8% of patients (2% = Grade 3). Monitor during treatment with FOTIVDA. For moderate to severe proteinuria, reduce the dose or interrupt treatment. Discontinue in patients who develop nephrotic syndrome.
Gastrointestinal (GI) Perforation including fatal cases, has been reported in patients receiving FOTIVDA. Monitor for symptoms of GI perforation or fistula formation periodically throughout treatment with FOTIVDA. Permanently discontinue FOTIVDA in patients who develop severe or life-threatening GI perforation.
Thyroid Dysfunction events were reported in 11% of patients (0.3% ≥ Grade 3). Monitor thyroid function before and during treatment with FOTIVDA.
Wound Healing Complications: Withhold FOTIVDA for at least 24 days prior to elective surgery and do not administer for at least 2 weeks after major surgery and until adequate wound healing is observed.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS) can occur with FOTIVDA. Evaluate for RPLS in patients presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue if signs or symptoms of RPLS occur.
Embryo-fetal Toxicity: FOTIVDA can cause fetal harm. Advise patients of the potential risk to a fetus, to avoid becoming pregnant and to use contraception during treatment and for one month after the last dose of FOTIVDA. Advise males with female partners of reproductive potential to use effective contraception during treatment and for one month after the last dose of FOTIVDA.
Allergic Reaction to Tartrazine: FOTIVDA 0.89 mg capsule contains FD&C Yellow No. 5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible patients.
ADVERSE REACTIONS
Common adverse reactions include fatigue/asthenia, hypertension, diarrhea, decreased appetite, nausea, dysphonia, hypothyroidism, cough, and stomatitis.
Serious adverse reactions include bleeding (3.5%), venous thromboembolism (3.5%), arterial thromboembolism (2.9%), acute kidney injury (2.3%), and hepatobiliary disorders (2.3%).
DRUG INTERACTIONS
Avoid coadministration with strong CYP3A4 inducers.
USE IN SPECIFIC POPULATIONS
Advise women not to breastfeed during treatment and for at least 1 month after the last dose.
The recommended dosage for patients with end-stage renal disease has not been established.
Reduce the FOTIVDA dose for patients with moderate hepatic impairment. The recommended dosage in patients with severe hepatic impairment has not been established.
To report SUSPECTED ADVERSE REACTIONS, contact AVEO Pharmaceuticals, Inc. at 1-833-FOTIVDA (1-833-368-4832) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information for FOTIVDA® (tivozanib).
